

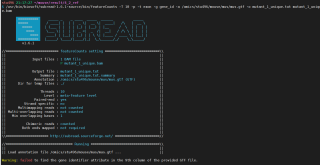
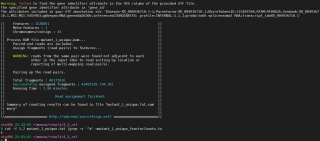
我在利用subread软件进行counts数计算的时候报了warning,但程序还能继续运行
导出txt文件后发现里面很乱,所有的结果都混在一行
想请教是哪一步出问题了 非常感谢

```shell
下面是使用的代码
/usr/bin/biosoft/subread-1.6.1-source/bin/featureCounts -T 10 -p -t exon -g gene_id -a /omics/stu496/mouse/mus/mus.gtf -o mutant_1_unique.txt mutant_1_unique.bam
/omics/stu496/mouse/mus/mus.gtf是之前在ncbi上下载的gff文件
mutant_1_unique.bam是用tophat对fastq文件比对后的bam文件
``






