vasp计算钙钛矿结构的声子谱时,出现虚频太多的情况,想问一下是计算出现了问题,还是高对称点的选择出现了问题
我计算的钙钛矿结构
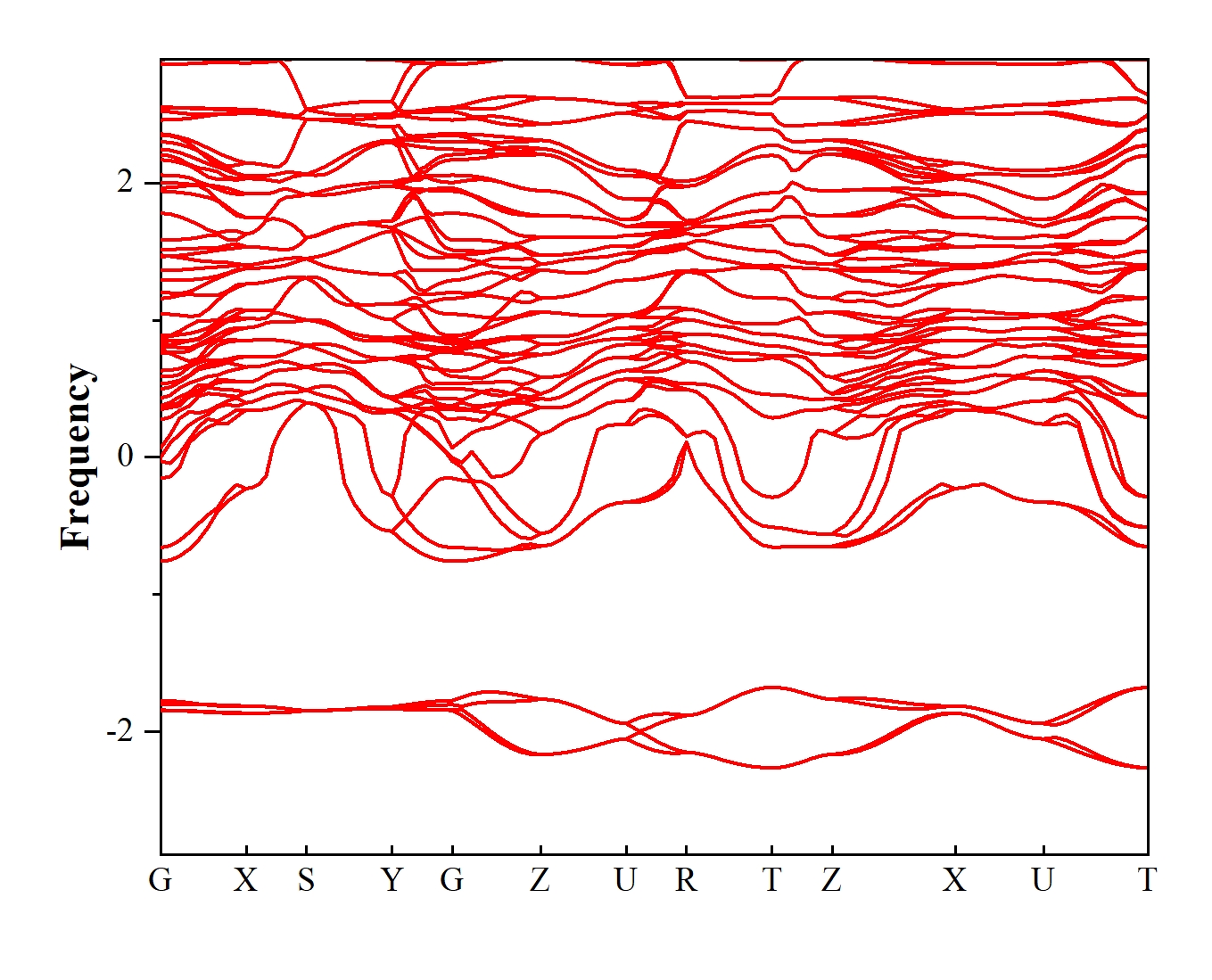
做的图

vasp计算钙钛矿结构的声子谱时,出现虚频太多的情况,想问一下是计算出现了问题,还是高对称点的选择出现了问题
 分享
分享
我曾经遇到过类似的问题,在使用VASP计算CH₃NH₃PbI₃钙钛矿结构声子谱时出现虚频过多的情况。这可能是由计算设置或结构本身的问题导致的,以下是几种可能的原因分析及解决方案:
核心思路:确保原子和晶胞充分弛豫,消除结构畸变引起的虚频。
操作步骤:
静态弛豫设置
EDIFF=1E-6 eV,力收敛阈值设为EDIFFG=-1E-3 eV/Å(注意负号表示收敛标准)。 IBRION=2或3,ISIF=3),确保晶格参数优化。 NELM=200),避免因未收敛导致的虚假力。 部分代码片段(INCAR文件):
IBRION = 2 ! 离子弛豫模式(2为标准共轭梯度法)
ISIF = 3 ! 同时弛豫原子和晶胞
EDIFF = 1E-6 ! 能量收敛阈值
EDIFFG = -1E-3 ! 力收敛阈值(绝对值为1E-3 eV/Å)
NELM = 200 ! 最大电子步次数
NSW = 100 ! 最大离子步次数
检查弛豫结果
VDW_POTENTIAL = TRUE或使用optB86b-vdW泛函),避免因弱相互作用描述不足导致结构畸变。核心思路:提高K点采样密度,选择合适的赝势,确保声子谱计算精度。
操作步骤:
加密K点网格
MONKHOST-PACK生成网格,例如MP = 8 8 8(原胞)或4 4 4(超胞),避免布里渊区采样不足。 部分代码片段(KPOINTS文件):
KPOINTS
0
Monkhorst-Pack
8 8 8 ! K点网格密度(根据体系大小调整)
0 0 0 ! 偏移量
更换赝势
PAW_PBE赝势(而非LDA),并确保I元素赝势包含足够的角动量态(如p态)。 核心思路:通过虚频的振动方向判断结构缺陷,针对性调整模型。
操作步骤:
vasp-phonopy或phonopy工具生成原子位移文件,观察虚频对应的振动模式。 推荐方案1(结构弛豫优化):
虚频过多通常源于结构未充分弛豫,严格的弛豫能从根本上消除几何畸变引起的虚假振动。优化后若残余力达标,声子谱中的虚频应显著减少。若仍存在少量虚频,可结合方案2进一步加密K点或更换赝势。
希望以上方案能帮你解决问题!若有具体计算结果或报错信息,可进一步留言讨论。请楼主采纳,如有问题请继续留言。
分享 创建了问题
5月19日
创建了问题
5月19日


