进行比对时一直提示fasta文件格式有问题

但是fasta文件从seqkit保存出来就是这样的格式没有更改过
求问如何解决/(ㄒoㄒ)/~~
修正之后还是一样/(ㄒoㄒ)/~~
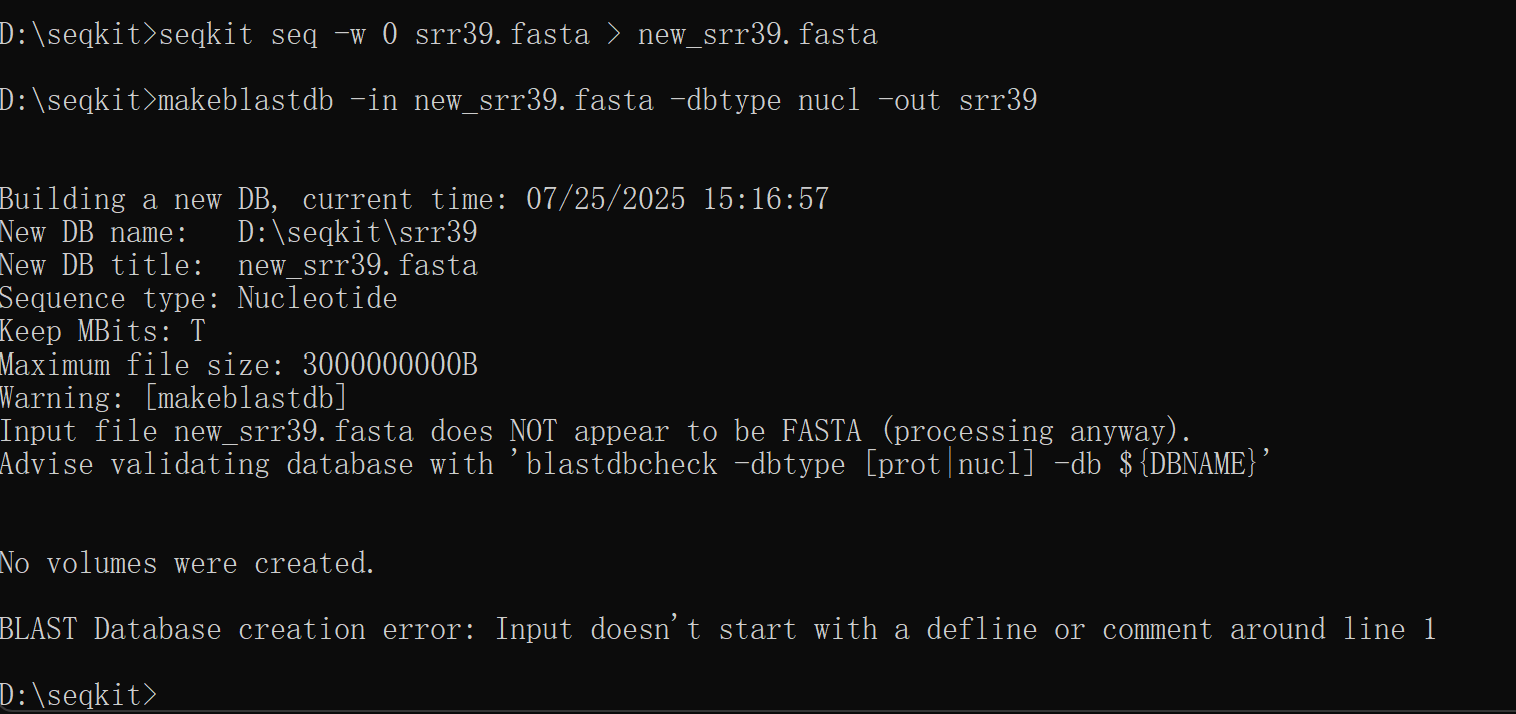

进行比对时一直提示fasta文件格式有问题
修正之后还是一样/(ㄒoㄒ)/~~
 分享
分享
 关注
关注让 “紫雾凌寒” 来帮你回答,本回答结合 DeepSeek
如果还有问题,请评论或点击头像私信,如果有帮助到您, 请关注、采纳、点赞
def check_fasta_format(fasta_file):
with open(fasta_file, 'r') as f:
lines = f.readlines()
for i, line in enumerate(lines):
if line.startswith('>'):
continue
else:
if not all(c in 'ACGTNacgtn-' for c in line.strip()):
return False
return True
fasta_file = 'your_file.fasta'
if check_fasta_format(fasta_file):
print('fasta文件格式正确')
else:
print('fasta文件格式有问题')
需要注意的是,以上代码只是一个简单的示例,实际应用中可能需要根据具体情况进行调整和完善。同时,也可以使用专门的生物信息学工具来更全面地检查和处理fasta文件格式。
希望以上解答对您有所帮助。如果您有任何疑问,欢迎在评论区提出。
分享 修改了问题
7月25日
创建了问题
7月25日
修改了问题
7月25日
创建了问题
7月25日

