GROMACS:为什么在使用gmx_mpi pdb2gmx -f 1_protein.pdb -o 1_protein.gro -p 1_protein.top -ignh -water tip3p -ff amber14sb -ter这个代码的时候不管换什么力场都会显示这个错误?
尝试过删掉OC1 OC2和都改成O都不对,应该怎么办啊?
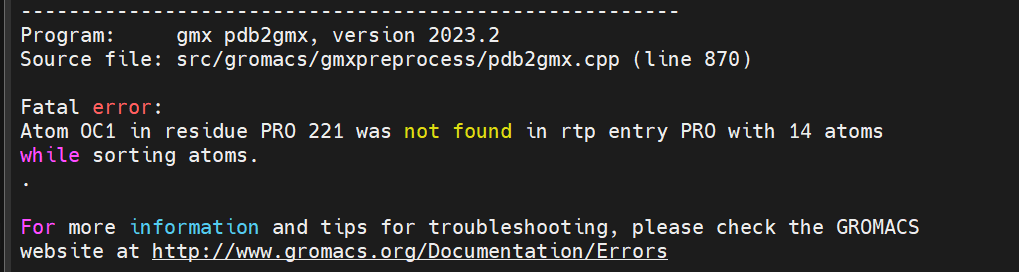

GROMACS:为什么在使用gmx_mpi pdb2gmx -f 1_protein.pdb -o 1_protein.gro -p 1_protein.top -ignh -water tip3p -ff amber14sb -ter这个代码的时候不管换什么力场都会显示这个错误?
尝试过删掉OC1 OC2和都改成O都不对,应该怎么办啊?
 分享
分享
下午好🌅🌅🌅
本答案参考通义千问
你遇到的 GROMACS 在使用 gmx_mpi pdb2gmx 命令时出现错误的问题,可能是由于以下几个原因导致的。以下是一个详细的分析和解决方案:
你执行的命令是:
gmx_mpi pdb2gmx -f 1_protein.pdb -o 1_protein.gro -p 1_protein.top -ignh -water tip3p -ff amber14sb -ter
这个命令的目的是将 .pdb 文件转换为 .gro 和 .top 文件,并选择力场为 amber14sb,水模型为 tip3p,同时忽略氢原子(-ignh)。
-ter 参数使用不当)。重点:确保 PDB 文件中没有非标准残基或原子名称。
1_protein.pdb,查看是否有以下内容:
OC1, OC2, OXT 等)HIS, GLU, ASP 等可能需要调整)OC1, OC2,可以将其重命名为 O 或 O1。OXT,可将其改为 O。注意: 不要随意删除原子,否则会影响模拟精度。
-ignore-modres 参数重点:避免因修饰残基(modres)导致的错误。
在 pdb2gmx 中添加参数:
gmx_mpi pdb2gmx -f 1_protein.pdb -o 1_protein.gro -p 1_protein.top -ignh -water tip3p -ff amber14sb -ter -ignore-modres
这会忽略所有修饰残基,避免因不支持的残基类型导致错误。
如果你发现某些原子名不一致,可以使用脚本或工具进行批量替换。
sed 替换 OC1 → Osed 's/OC1/O/g' 1_protein.pdb > 1_protein_cleaned.pdb
然后用新文件运行命令:
gmx_mpi pdb2gmx -f 1_protein_cleaned.pdb -o 1_protein.gro -p 1_protein.top -ignh -water tip3p -ff amber14sb -ter
-no-water 选项(测试模式)重点:先测试蛋白质部分是否能成功生成拓扑。
你可以暂时不加水模型,看是否能正常运行:
gmx_mpi pdb2gmx -f 1_protein.pdb -o 1_protein.gro -p 1_protein.top -ignh -ff amber14sb -ter
如果这一步成功,说明问题可能出在水模型或力场组合上。
重点:确认 GROMACS 安装正确,且版本兼容。
gmx_mpi --version
-force-field 指定更通用的力场(可选)重点:尝试使用 charmm36 或 oplsaa 力场,看看是否能解决问题。
gmx_mpi pdb2gmx -f 1_protein.pdb -o 1_protein.gro -p 1_protein.top -ignh -water tip3p -ff charmm36 -ter
-ignore-modres 忽略修饰残基。charmm36)测试。gmx_mpi pdb2gmx -f 1_protein_cleaned.pdb -o 1_protein.gro -p 1_protein.top -ignh -water tip3p -ff amber14sb -ter -ignore-modres
如果你仍然无法解决,请提供以下信息以便进一步诊断:
1_protein.pdb 的前几行(例如 10 行左右);我可以帮你进一步分析!
分享 创建了问题
8月2日
创建了问题
8月2日

