开始运行后,第一步就出错了,一共设置了5000步,没跑完。然后pmemd就失败了,看了nohup.out里,显示如图,怎么解决呢?
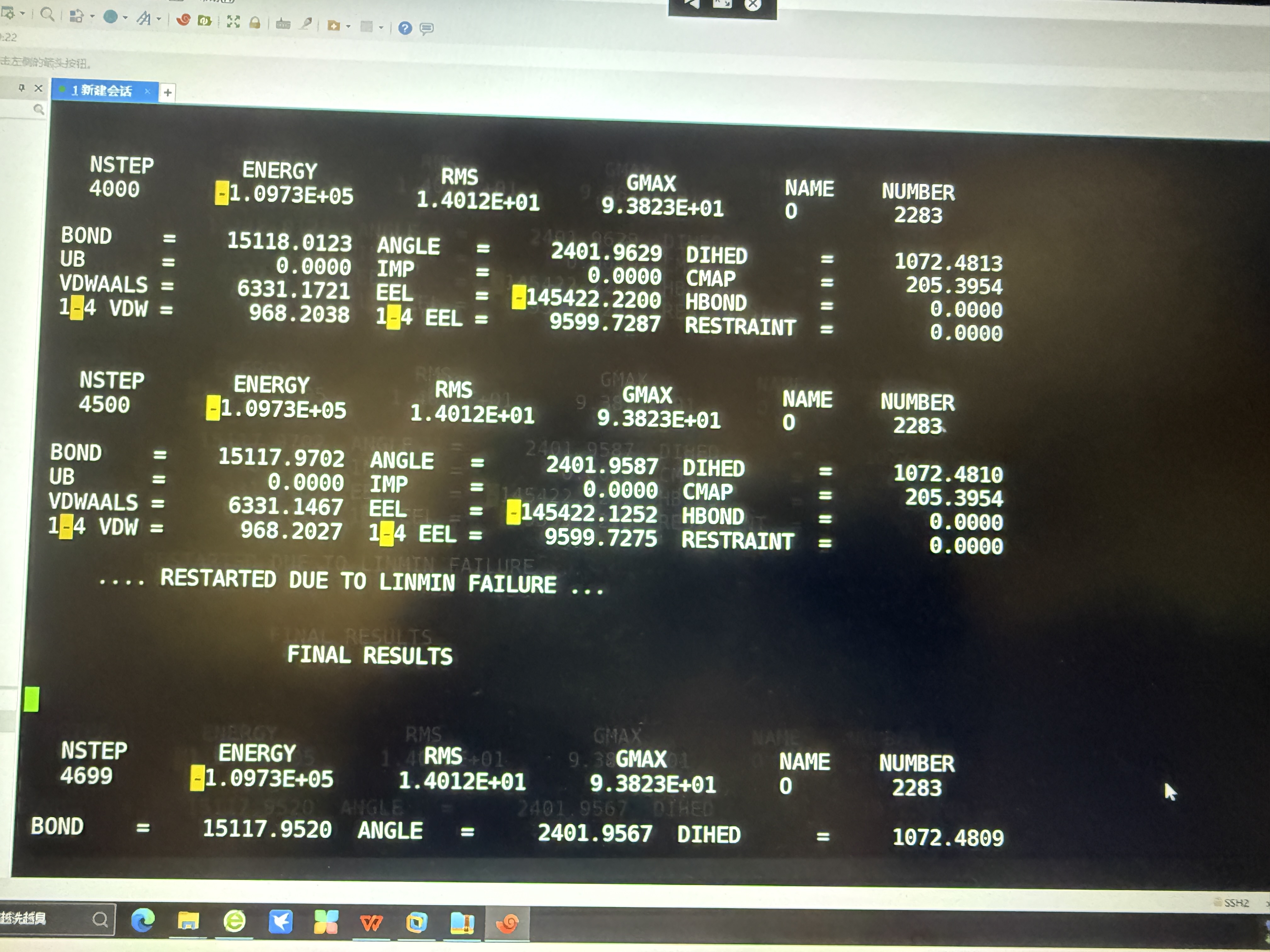
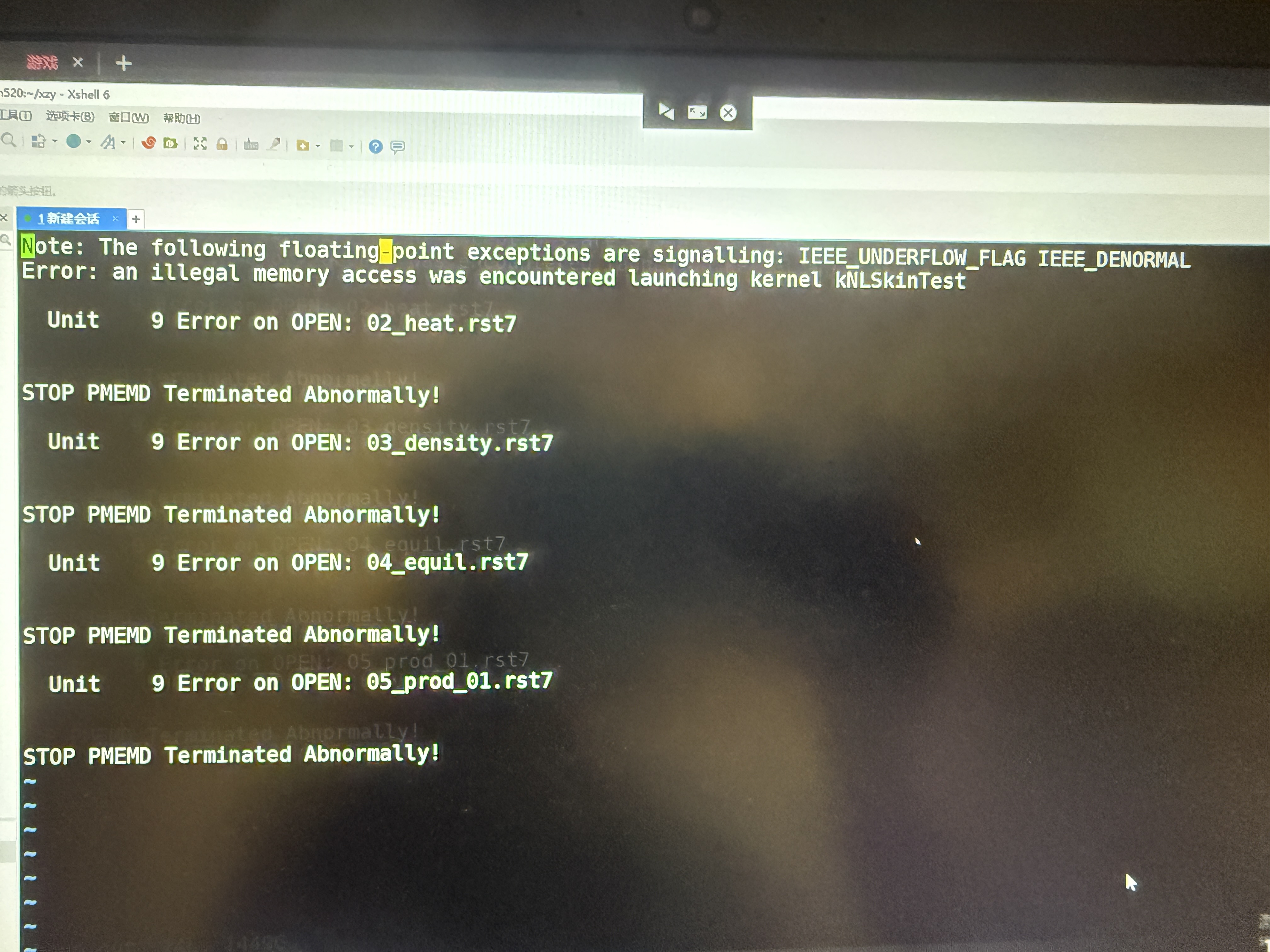

开始运行后,第一步就出错了,一共设置了5000步,没跑完。然后pmemd就失败了,看了nohup.out里,显示如图,怎么解决呢?
 分享
分享
下午好🌅🌅🌅
本答案参考通义千问
根据你描述的问题,pmemd.cuda 在运行过程中失败,且只运行了5000步就出错,同时在 nohup.out 中有错误信息。我们可以从以下几个方面来排查和解决这个问题:
由于你提到“看了 nohup.out 里显示如图”,但没有提供图片,我们假设你看到的是以下常见错误类型之一:
建议:请将
nohup.out中的完整错误信息复制粘贴过来,这样可以更精准地定位问题。
CUDA error: out of memory 或类似提示# 原命令(可能超出内存)
pmemd.cuda -O -i in.prm -o out.mdout -p topol.top -c conf.gro -r restart.rst7 -x traj.xtc
# 修改为使用较小的 GPU 模型(如使用单个 GPU)
mpirun -np 1 pmemd.cuda -O -i in.prm -o out.mdout -p topol.top -c conf.gro -r restart.rst7 -x traj.xtc
注意:如果使用多 GPU,确保每块 GPU 的显存足够。
nohup.out 中出现 Segmentation fault (core dumped) 或 Bus error.top、坐标文件 .gro)pmemd 而不是 pmemd.cuda(测试是否是 CUDA 相关问题)# 使用 CPU 版本进行测试
pmemd -O -i in.prm -o out.mdout -p topol.top -c conf.gro -r restart.rst7 -x traj.xtc
如果 CPU 版本运行成功,则可能是 CUDA 驱动或编译问题。
NaN 或 Invalid energy valuein.prm 文件中的参数设置(如 ntt=1, tautp=0.1 等)in.prm 文件片段:&cntrl
nstlim = 5000,
dt = 0.002,
ntt = 1,
tautp = 0.1,
temp0 = 300.0,
tautp = 0.1,
ntp = 1,
pres0 = 1.0,
ntc = 2,
ntf = 2,
cut = 10.0,
imin = 0,
ntpr = 100,
ntwx = 100,
ntwr = 500,
/
重点:确保
dt设置合理,避免数值不稳定。
Cannot open file ...ls -l topol.top conf.gro restart.rst7
查看 nohup.out 中的具体错误信息
如果能提供具体错误内容,可直接定位问题。
使用 CPU 版本 pmemd 测试
排除是否为 CUDA 相关问题。
检查输入文件格式是否正确
如
.top,.gro,.rst7是否符合要求。
减少模拟步数或规模
例如从 5000 步改为 1000 步,看是否能运行。
检查显卡驱动和 CUDA 版本兼容性
确保安装了与
pmemd.cuda兼容的 CUDA 驱动。
更新 Amber 工具链
推荐使用最新版本(如 Amber22 或更高)。
#!/bin/bash
# 使用单 GPU 运行 pmemd.cuda
export AMBERHOME=/path/to/amber22
source $AMBERHOME/bin/amber.sh
# 设置环境变量
export OMP_NUM_THREADS=1
export CUDA_VISIBLE_DEVICES=0
# 运行 pmemd.cuda
mpirun -np 1 pmemd.cuda -O -i in.prm -o out.mdout -p topol.top -c conf.gro -r restart.rst7 -x traj.xtc
重点:确保
CUDA_VISIBLE_DEVICES和mpirun参数配置正确。
| 问题 | 可能原因 | 解决方法 |
|------|----------|----------|
| CUDA 内存不足 | 显存不够 | 减少 GPU 数量,优化输入结构 |
| 程序崩溃 | 输入文件错误 | 检查 .top, .gro 文件 |
| 参数设置错误 | 时间步长过大 | 调整 dt 为 2 fs |
| 文件路径错误 | 权限或路径错误 | 检查文件存在性和权限 |
如果你能提供更多关于 nohup.out 的具体错误信息,我可以进一步帮你分析。欢迎继续提问!
分享 【Amber 安装教程】入坑Amber须知 Pmemd vs. AmberTools 区别 AmberTools 能做什么 AmberTools 核心功能 新手安装Amber 以及AmberTools
【Amber 安装教程】入坑Amber须知 Pmemd vs. AmberTools 区别 AmberTools 能做什么 AmberTools 核心功能 新手安装Amber 以及AmberTools 创建了问题
9月15日
创建了问题
9月15日
