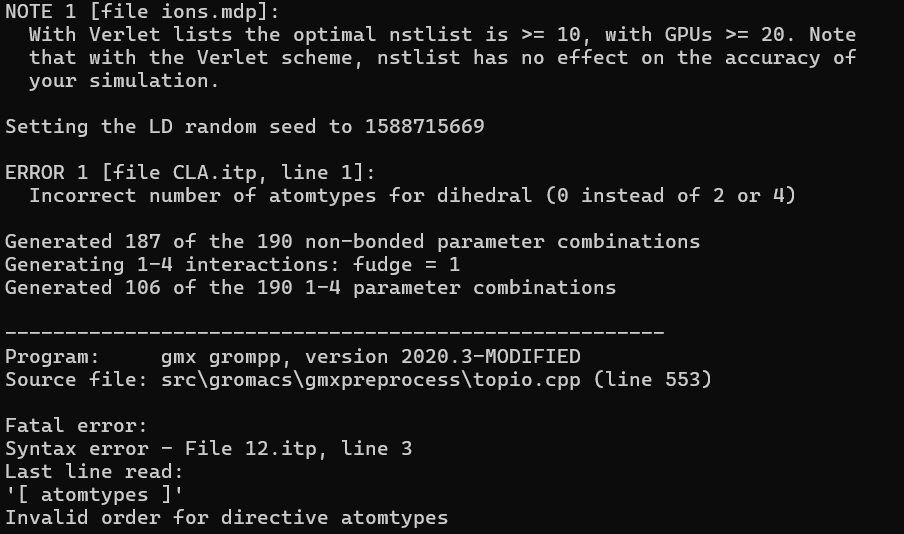
请问利用gromacs进行药物小分子跨磷脂双分子层的分子动力学模拟,出现这种报错该如何解决?

请问利用gromacs进行药物小分子跨磷脂双分子层的分子动力学模拟,出现这种报错该如何解决?
 分享
分享
阿里嘎多学长整理AIGC生成,因移动端显示问题导致当前答案未能完全显示,请使用PC端查看更加详细的解答过程
解决GROMACS求解错误的方法
根据你的问题描述,GROMACS求解错误可能是由于各种原因引起的,以下是一些常见的解决方法:
核心代码
# 检查输入文件
gmx check -f your_file.gro
# 检查参数设置
gmx grompp -f your_file.mdp -c your_file.gro
# 检查系统的稳定性
gmx energy -f your_file.gro -o energy.xvg
# 检查计算资源
gmx mdrun -v -s your_file.tpr -o your_file.trr
如果以上方法仍然无法解决问题,请提供更多详细信息和错误日志,我将尽力帮助你解决问题。
分享 创建了问题
10月25日
创建了问题
10月25日


