在windows虚拟的ubantu系统中安装生信比对STAR软件,运行过程中出现奇怪BUG
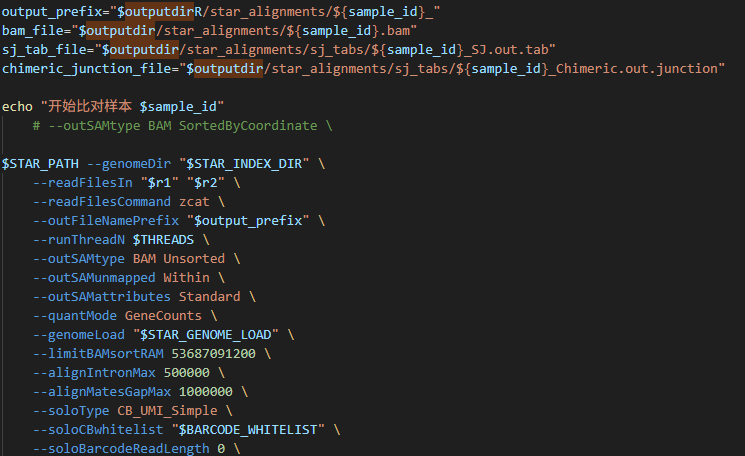
如图片所示,在定义变量时我误加入了一个大写R字母,脚本运行正常,只是结果文件存储位置并非预期的位置(F盘)结果保存文件位置如下图:
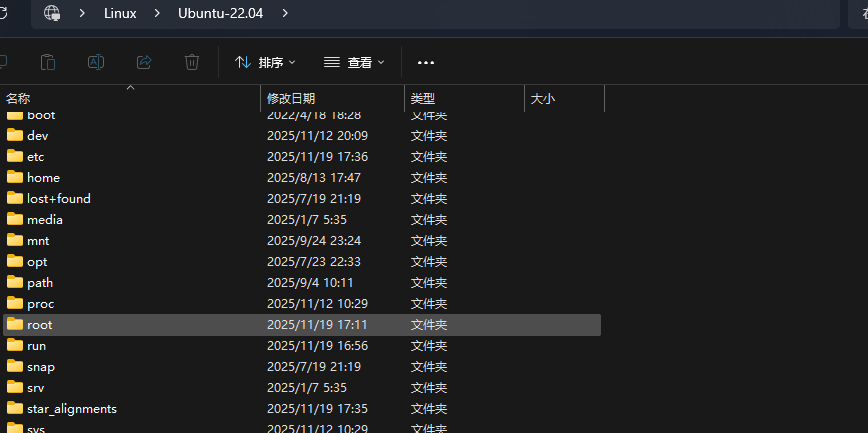
而当我将这个BUG修复时,运行脚本会出现以下报错

百思不得其解……

在windows虚拟的ubantu系统中安装生信比对STAR软件,运行过程中出现奇怪BUG
如图片所示,在定义变量时我误加入了一个大写R字母,脚本运行正常,只是结果文件存储位置并非预期的位置(F盘)结果保存文件位置如下图:
而当我将这个BUG修复时,运行脚本会出现以下报错
百思不得其解……
 分享
分享
 关注
关注让【宝码香车】来帮您解答,本回答参考DeepSeek编写,并整理提供。
如果答案能帮助您,请采纳,非常感谢!
.
每日一句正能量,让它带给您好心情:即使走得很慢,只要不后退,就是在前进。
.
根据您提供的信息,这是一个典型的文件系统兼容性问题。让我为您详细分析问题和提供解决方案。
从图片3的报错信息可以看出核心问题:
FATAL ERROR: could not create FIFO file /mnt/f/scRNA_results/star_alignments/L_MASH_GEX_1__STARtmp/tmp.fifo.read1
SOLUTION: check if run directory supports FIFO files.
根本原因:STAR软件在运行过程中需要创建FIFO(命名管道)文件,但Windows的NTFS/FAT文件系统不支持FIFO文件。
当您使用错误的变量定义时:
output_prefix=" R/star_alignments/${sample id} "
STAR实际上将结果输出到了Ubuntu系统根目录下的R/文件夹,而不是Windows共享的/mnt/f/目录。Linux原生文件系统(如ext4)支持FIFO,所以能够正常运行。
--outTmpDir参数(推荐)将临时目录指向Linux原生分区:
# 在home目录或其他Linux分区创建临时目录
mkdir -p ~/star_tmp
# 修改STAR命令,添加outTmpDir参数
$STAR_PATH --genomeDir "$STAR_INDEX_DIR" \
--readFilesIn "$r1" "$r2" \
--readFilesCommand zcat \
--outFileNamePrefix "$output_prefix" \
--runThreadN $THREADS \
--outSAMtype BAM Unsorted \
--outSAMunmapped Within \
--outSAMattributes Standard \
--quantMode GeneCounts \
--genomeLoad "$STAR_GENOME_LOAD" \
--limitBAMsortRAM 53687691260 \
--alignIntronMax 500000 \
--alignMatesGapMax 1060000 \
--soloType CB_UMI_Simple \
--soloCBwhitelist "$BARCODE_WHITELIST" \
--soloBarcodeReadLength 8 \
--outTmpDir ~/star_tmp/${sample_id}
将数据复制到Ubuntu的家目录中处理:
# 将数据从/mnt/f/复制到home目录
cp -r /mnt/f/scRNA_results ~/scRNA_results
# 修改脚本中的路径指向home目录
output_prefix="$HOME/scRNA_results/star_alignments/${sample_id}"
如果使用VirtualBox/VMware,确保共享文件夹以支持Linux特性的方式挂载。
#!/bin/bash
# 定义变量(修正版本)
sample_id="L_MASH_GEX_1"
output_prefix="/mnt/f/scRNA_results/star_alignments/${sample_id}"
bam_file="/mnt/f/scRNA_results/star_alignments/${sample_id}.bam"
sj_tab_file="/mnt/f/scRNA_results/star_alignments/sj_tabs/${sample_id}_SJ.out.tab"
chimeric_junction_file="/mnt/f/scRNA_results/star_alignments/sj_tabs/${sample_id}_Chimeric.out.junction"
# 创建必要的目录
mkdir -p /mnt/f/scRNA_results/star_alignments/sj_tabs
mkdir -p ~/star_tmp/${sample_id}
echo "开始比对样本 $sample_id"
$STAR_PATH --genomeDir "$STAR_INDEX_DIR" \
--readFilesIn "$r1" "$r2" \
--readFilesCommand zcat \
--outFileNamePrefix "$output_prefix" \
--runThreadN $THREADS \
--outSAMtype BAM Unsorted \
--outSAMunmapped Within \
--outSAMattributes Standard \
--quantMode GeneCounts \
--genomeLoad "$STAR_GENOME_LOAD" \
--limitBAMsortRAM 53687691260 \
--alignIntronMax 500000 \
--alignMatesGapMax 1060000 \
--soloType CB_UMI_Simple \
--soloCBwhitelist "$BARCODE_WHITELIST" \
--soloBarcodeReadLength 8 \
--outTmpDir ~/star_tmp/${sample_id}
# 处理完成后清理临时目录
rm -rf ~/star_tmp/${sample_id}
使用方案1应该能解决您的问题。如果仍有问题,请考虑将整个工作流程迁移到Linux原生分区。
分享 系统已结题
11月27日
系统已结题
11月27日 已采纳回答
11月19日
创建了问题
11月19日
已采纳回答
11月19日
创建了问题
11月19日

