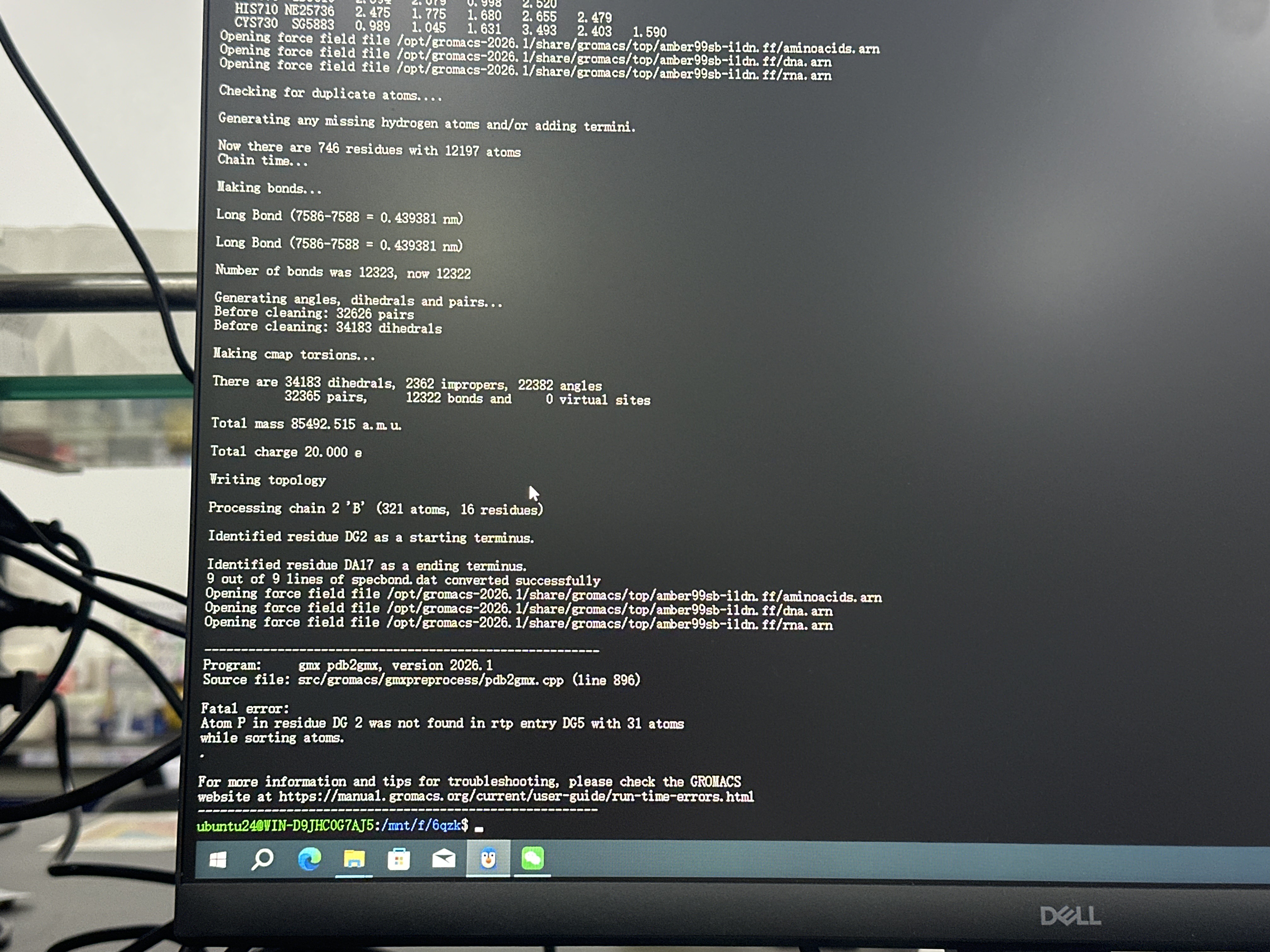
谁能帮我看一下用Linux里面跑GROMACS一直报错,蛋白质是没问题的但是DNA一直显示有问题是为什么,而且已经改了很多遍了

 分享
分享
晚上好🌙🌙🌙
本答案参考通义千问
在使用 GROMACS 时,如果蛋白质部分可以正常运行,但 DNA 部分一直报错,可能是由于以下原因导致的。下面我将详细分析可能的原因,并提供解决方案。
DNA 的拓扑文件(如 .top 或 .itp 文件)中可能存在语法错误或不兼容的定义。
.top 文件中是否正确包含了 DNA 的 include 语句。gmx check 工具验证拓扑文件是否有问题。gmx check -f your_system.top
重点: 拓扑文件中的每一行都必须严格符合 GROMACS 的格式要求。
GROMACS 对原子名称和类型有严格的定义,如果 DNA 中的原子名称或类型与标准库(如 ffnonbonded.itp 或 dna.itp)不一致,会导致错误。
gmx editconf 或 gmx pdb2gmx 检查并修正原子名称。.pdb),请确保原子名称与 GROMACS 标准库一致。gmx pdb2gmx -f dna.pdb -o dna_processed.gro -water spc
重点: 使用
pdb2gmx自动处理 DNA 可以避免许多格式问题。
DNA 和蛋白质之间可能会有静电、范德华等非键作用,若这些参数未正确设置,也可能导致模拟失败。
.top 文件中检查是否包含了 DNA 与蛋白质之间的 pairtypes 或 exclusions。include 语句正确指向 DNA 的力场文件(如 dna.itp)。#include "dna.itp"
重点: DNA 的力场文件需要与蛋白质使用的力场(如
protein.itp)兼容。
DNA 通常是带负电的,如果其电荷未正确计算,可能导致系统整体电荷不平衡,从而引发错误。
gmx charge 检查系统电荷。gmx pdb2gmx 正确分配。gmx charge -f system.gro -n index.ndx
重点: 如果电荷总和不为零,建议添加适当的离子平衡。
有时候,GROMACS 报错是由于路径或文件名错误导致的,尤其是 DNA 相关文件没有被正确加载。
.top 文件中是否包含正确的 DNA 文件路径。dna.itp、solvent.itp)都在当前目录下或在 GMXDATA 路径中。不同版本的 GROMACS 对某些力场或拓扑格式的支持可能不同。
检查拓扑文件格式
gmx check 验证 .top 文件是否合法。include 语句正确。使用 pdb2gmx 处理 DNA 坐标文件
验证系统电荷
gmx charge 检查电荷是否平衡。检查 DNA 与蛋白质之间的相互作用
确认文件路径和命名正确
尝试更新或降级 GROMACS
.top 文件)如果你的 .top 文件类似如下结构,请检查是否缺少 include 或格式错误:
#include "forcefield.itp"
#include "dna.itp"
[ moleculetype ]
; name nrexcl
DNA 3
[ atoms ]
; id type resnr atomname cgnr charge mass
1 C1 1 C1 1 0.0 12.011
...
[ bonds ]
...
重点: 确保 DNA 的
moleculetype和atoms部分完整且格式正确。
如果你能提供具体的错误信息或 .top 文件内容,我可以更准确地帮你定位问题!
分享 【Amber 安装教程】入坑Amber须知 Pmemd vs. AmberTools 区别 AmberTools 能做什么 AmberTools 核心功能 新手安装Amber 以及AmberTools
【Amber 安装教程】入坑Amber须知 Pmemd vs. AmberTools 区别 AmberTools 能做什么 AmberTools 核心功能 新手安装Amber 以及AmberTools 创建了问题
4月1日
创建了问题
4月1日
