从GEO下载的环状RNA测序数据,数据类型是reads数,我该如何将其转化为counts、CPM或者FPKM数据?
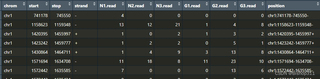

从GEO下载的环状RNA测序数据,数据类型是reads数,我该如何将其转化为counts、CPM或者FPKM数据?
 分享
分享
Bishop311 上午好☀️☀️☀️️
本答案参考ChatGPT-3.5
要将环状RNA二代测序得到的reads数转化为counts、CPM或FPKM数据,可以按照以下步骤进行:
将reads数转换为counts数据:
将counts数据转换为CPM(Counts Per Million)数据:
将counts数据转换为FPKM(Fragments Per Kilobase Million)数据:
以下是一个简单的示例代码,演示如何将reads数转换为counts和FPKM数据:
import pandas as pd
# 假设读取的数据存储在一个名为"data.csv"的CSV文件中
data = pd.read_csv("data.csv", delimiter="\t")
# 计算每个样本的标准化因子
normalization_factors = data.sum(axis=1)
# 将每个样本的reads数除以标准化因子得到counts数据
counts_data = data.iloc[:, 4:].div(normalization_factors, axis=0)
# 计算每个样本的总counts数
total_counts = counts_data.sum()
# 计算每个基因的长度
# 这里假设基因长度存储在一个名为"gene_lengths.csv"的CSV文件中
gene_lengths = pd.read_csv("gene_lengths.csv")
# 根据每个样本的总counts数,基因长度和每百万的缩放因子计算出每个基因的FPKM值
fpkm_data = counts_data.div((total_counts / 1e6), axis=1).div(gene_lengths["length"], axis=0)
# 输出counts和FPKM数据
counts_data.to_csv("counts_data.csv", index=False)
fpkm_data.to_csv("fpkm_data.csv", index=False)
请注意,这只是一个示例代码,具体的代码实现可能因数据格式和处理要求而有所不同。要根据自己的数据进行修改和调整,以适应特定的数据和分析需求。
分享 系统已结题
4月3日
系统已结题
4月3日 创建了问题
3月26日
创建了问题
3月26日

