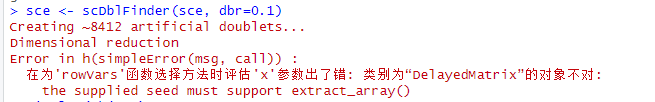
求./助,我首先将单细胞数据进行标准化和降维处理,然后用scDblFinder去除双胞,但是一直报错。
object <- Read10X('E:\\R\\brain\\MER2\\10K3') #读取单细胞数据
object <- CreateSeuratObject(counts = object, project = "brain_1", min.cells = 3, min.features = 200) #创建seurat对象
object[["percent.mt"]] <- PercentageFeatureSet(object, pattern = "ATP6|ATP8|COX1|COX2|COX3|CYTB|ND1|ND2|ND3|ND4|ND4L|ND5|ND6")
if (sum(object@meta.data$percent.mt) == 0){
object@meta.data$percent.mt[1] <- 0.000001
} ## 计算线粒体基因百分比,0值补成非0的极小数
object <- subset(object, subset = nFeature_RNA > 200 & nFeature_RNA < 6000 & nCount_RNA < 20000 & percent.mt < 5) # 200 < nFeature_RNA(单个细胞总基因数) < 6000; # nCount_RNA(单个细胞总表达量数) < 20000; # percent.mt(单个细胞内线粒体基因比例) < 5
# 进行seurat流程的预处理,三步标准化。
object <- NormalizeData(object)
object <- FindVariableFeatures(object, selection.method = "vst", nfeatures = 2000)
object <- ScaleData(object)
object <- RunPCA(object)
object <- RunUMAP(object, dims = 1:20)
object <- FindNeighbors(object, reduction = "pca", dims = 1:20)
object <- FindClusters(object, resolution = 0.5)
sce <- as.SingleCellExperiment(object)

sce <- scDblFinder(sce, dbr=0.1)