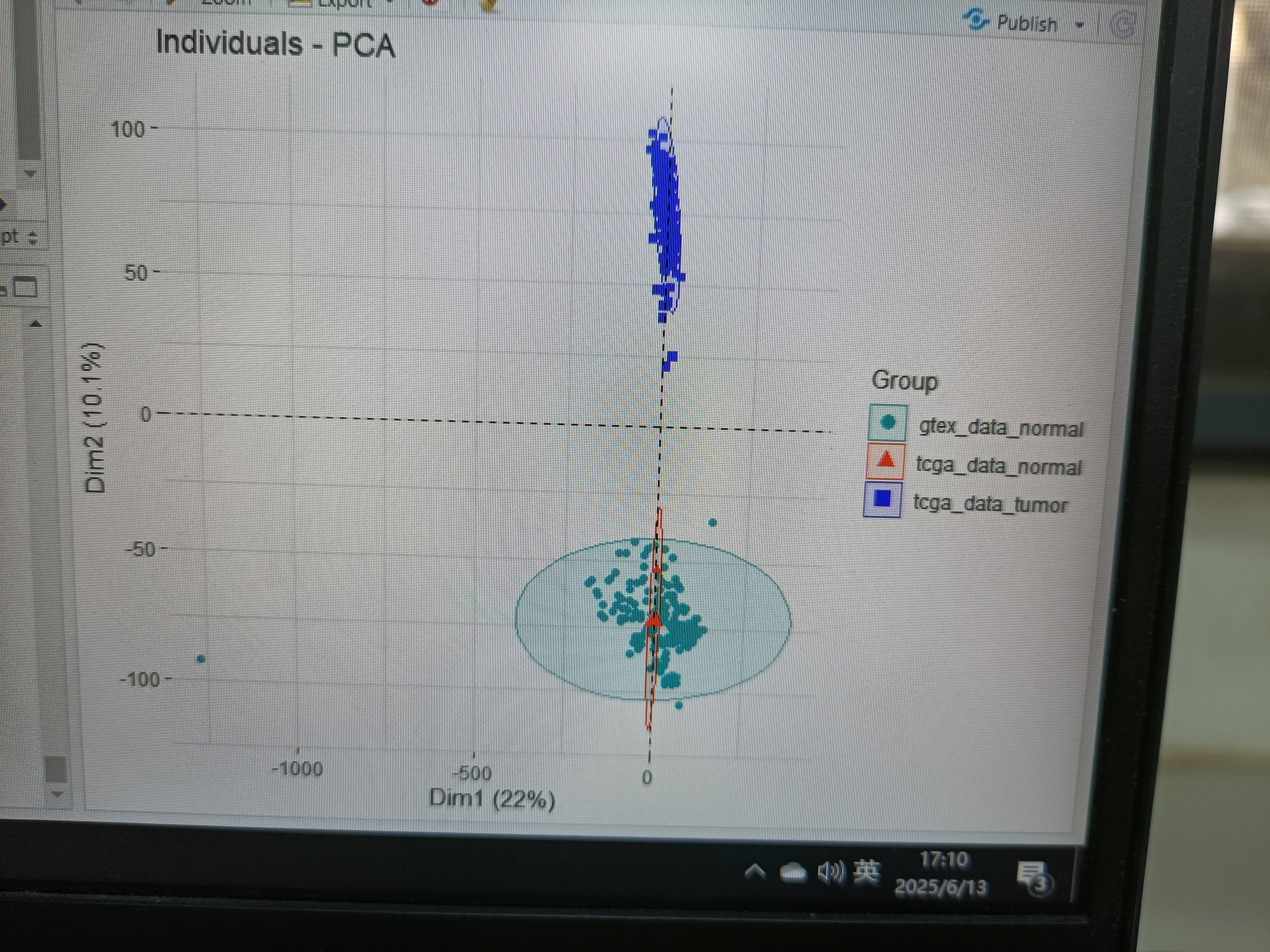
我取了175个tcga数据以及随机取了gtex170个正常样本,合并出来这个样子,咋办啊

 分享
分享
阿里嘎多学长整理AIGC生成,因移动端显示问题导致当前答案未能完全显示,请使用PC端查看更加详细的解答过程
你想要将TCGA和GTEx数据合并,去批次,找到差异基因,然后进行差异分析。你的数据包括175个TCGA样本和170个GTEx正常样本。
首先,需要注意的是,TCGA和GTEx数据的格式和内容可能不同,需要进行数据预处理和合并。下面是一个可能的解决方案:
readr包读取TCGA和GTEx数据,转换为数据框格式。dplyr包合并数据框,使用inner_join或full_join函数根据样本ID或其他共同特征合并数据。limma包进行差异基因分析,选择合适的统计方法和参数。ggplot2包进行可视化,展示差异基因的结果。以下是一个简单的示例代码:
library(readr)
library(dplyr)
library(limma)
library(ggplot2)
# 读取TCGA数据
tcga_data <- read_csv("tcga_data.csv")
# 读取GTEx数据
gtx_data <- read_csv("gtx_data.csv")
# 合并数据
merged_data <- inner_join(tcga_data, gtx_data, by = "sample_id")
# 进行差异基因分析
design <- model.matrix(~0 + factor(merged_data$group))
fit <- lmFit(merged_data$expression, design)
ebayes <- eBayes(fit)
# 选择差异基因
diff_genes <- rownames(ebayes)[ebayes$adj.P.Val < 0.05]
# 可视化差异基因
ggplot(merged_data[diff_genes, ], aes(x = group, y = expression)) +
geom_boxplot() +
theme_classic()
请注意,这只是一个简单的示例代码,实际情况中可能需要更多的数据预处理和分析步骤。
分享 创建了问题
6月13日
创建了问题
6月13日


