晚上好🌙🌙🌙
本答案参考通义千问
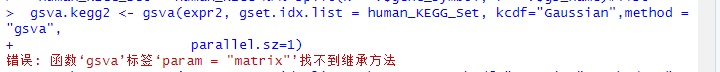
在使用 GSVA 进行通路分析时,出现“找不到继承方法”(no applicable method for 'gsva' applied to an object of class "matrix") 的错误,通常是因为 expr2 的数据类型不正确或 gset.idx.list 的格式不符合 GSVA 的要求。
✅ 问题原因分析:
expr2 不是矩阵:虽然你用了 as.matrix(expr2),但可能 expr2 本身不是矩阵或者数据框,导致无法正确传入 GSVA 函数。gset.idx.list 格式不正确:GSVA 要求 gset.idx.list 是一个 列表(list),其中每个元素是一个 基因符号的向量。你目前的 human_KEGG_Set 可能不是一个标准的基因集合列表。- GSVA 方法调用方式不正确:
gsva() 的参数需要符合其函数定义,比如 expr 应该是表达矩阵,gset.idx.list 是基因集合列表。
🔧 解决方案:
1. 确保 expr2 是一个正确的表达矩阵
# 检查 expr2 是否为矩阵
class(expr2)
# 如果不是矩阵,转换为矩阵
expr2 <- as.matrix(expr2)
重点:确保 expr2 是一个以基因名为行名、样本为列名的表达矩阵。
2. 正确构建 gset.idx.list
你的代码中:
human_KEGG_Set = human_KEGG %>% split(x = .$gene_symbol, f = .$gs_name)
这会将 human_KEGG 按照 gs_name 分组,生成一个 list,其中每个元素是一个 data.frame(包含 gene_symbol 列)。但 GSVA 需要的是每个元素是一个 字符向量(即基因名列表),而不是数据框。
修改后代码如下:
# 将每个基因集合转换为字符向量
human_KEGG_Set <- human_KEGG %>%
split(.$gs_name) %>%
lapply(function(x) x$gene_symbol)
重点:gset.idx.list 必须是一个 list,每个元素是 character 类型的基因名向量。
3. 使用正确的 GSVA 函数调用方式
GSVA 提供了多种方法(如 "gsva" 和 "ssgsea"),建议使用 gsva::gsva() 或 gsva::ssgsea()。
完整修正后的代码如下:
library(gsva)
library(dplyr)
library(msigdbr)
# 确保 expr2 是矩阵
expr2 <- as.matrix(expr2)
# 获取 KEGG 基因集
human_KEGG <- msigdbr(species = "Homo sapiens",
category = "C2",
subcategory = "CP:KEGG_LEGACY") %>%
dplyr::select(gs_name, gene_symbol)
# 构建基因集合列表
human_KEGG_Set <- human_KEGG %>%
split(.$gs_name) %>%
lapply(function(x) x$gene_symbol)
# 执行 GSVA 分析
gsva_result <- gsva(expr2, gset.idx.list = human_KEGG_Set, kcdf = "Gaussian", method = "gsva")
# 查看结果
head(gsva_result)
📌 总结关键点:
expr2 必须是矩阵,且行名是基因名,列名是样本名。gset.idx.list 必须是 list,每个元素是 character 向量。- GSVA 函数调用需符合其参数规范,推荐使用
gsva::gsva() 或 gsva::ssgsea()。
✅ 示例输出(可选):
# 查看前几行结果
head(gsva_result)
如果一切正常,你应该看到一个矩阵,其中行是通路名称,列是样本,值是 GSVA 得分。
如有其他报错,请提供完整的错误信息,我可以进一步帮你定位问题。