晚上好🌙🌙🌙
本答案参考通义千问
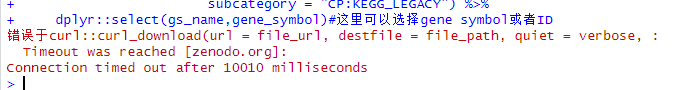
在使用 msigdbr 包加载基因集时,如果遇到超时(timeout)错误,通常是因为网络连接不稳定、服务器响应缓慢或数据量过大导致请求失败。以下是详细的解决方案和优化建议:
一、问题原因分析
- 网络连接不稳定:
msigdbr 从外部源(如 MSigDB)获取数据,若网络延迟较高,容易出现超时。 - 数据量过大:某些基因集包含大量基因,可能导致请求时间过长。
- 默认参数设置不合理:可能没有对请求进行合理的超时控制。
二、解决方法
1. 增加超时限制
可以通过修改 httr 的默认超时时间来避免因等待太久而中断请求。
options(httr_oauth_cache = FALSE)
options(timeout = 600) # 将超时时间设为600秒(10分钟)
注意: 这个设置需要在运行 msigdbr 之前设置。
2. 使用本地缓存(推荐)
为了避免每次都从网络下载数据,可以使用 msigdbr 的缓存功能,将数据保存到本地,后续直接读取。
# 设置缓存路径(可选)
cache_path <- "~/.msigdb"
dir.create(cache_path, showWarnings = FALSE)
# 获取基因集并缓存
human_KEGG <- msigdbr(
species = "Homo sapiens",
category = "C2",
subcategory = "CP:KEGG_LEGACY",
cache = TRUE,
cache_dir = cache_path
) %>%
dplyr::select(gs_name, gene_symbol)
human_KEGG_Set <- split(human_KEGG$gene_symbol, human_KEGG$gs_name)
优点: 第一次加载后,后续使用会非常快,且不会受网络影响。
3. 分页加载数据(适用于大规模数据)
如果数据量特别大,可以分批次加载,避免一次性请求过多数据。
library(msigdbr)
library(dplyr)
# 定义分页参数
page_size <- 500
total_pages <- 10
# 存储所有结果
all_gs <- list()
for (i in 1:total_pages) {
page_data <- msigdbr(
species = "Homo sapiens",
category = "C2",
subcategory = "CP:KEGG_LEGACY",
page = i,
page_size = page_size
) %>%
select(gs_name, gene_symbol)
all_gs[[i]] <- page_data
}
# 合并所有分页结果
human_KEGG <- bind_rows(all_gs)
human_KEGG_Set <- split(human_KEGG$gene_symbol, human_KEGG$gs_name)
说明: 需要根据实际数据量调整 total_pages 和 page_size。
4. 检查网络环境与代理设置
如果你在中国大陆,可能会因为网络原因导致无法访问 MSigDB,可以尝试以下方法:
- 使用国内镜像站点(如清华源);
- 设置 HTTP 代理(如使用
Sys.setenv(http_proxy = "http://your.proxy:port"))。
5. 替代方案:使用本地数据库或手动下载
如果上述方法仍然无法解决问题,可以考虑:
-
手动下载 MSigDB 数据库:
- 访问 MSigDB 官网
- 下载 KEGG 基因集文件(如
c2.cp.kegg.v7.5.1.entrez.gmt)
-
使用 gseapy 或 clusterProfiler 加载本地文件:
library(gseapy)
kegg_gmt <- gseapy.get_gmt("path/to/c2.cp.kegg.v7.5.1.entrez.gmt")
优点: 完全脱离网络依赖,适合离线环境。
三、完整代码示例(推荐方式)
# 设置超时时间
options(timeout = 600)
# 设置缓存路径
cache_path <- "~/.msigdb"
dir.create(cache_path, showWarnings = FALSE)
# 获取基因集并缓存
human_KEGG <- msigdbr(
species = "Homo sapiens",
category = "C2",
subcategory = "CP:KEGG_LEGACY",
cache = TRUE,
cache_dir = cache_path
) %>%
dplyr::select(gs_name, gene_symbol)
# 转换为基因集列表
human_KEGG_Set <- split(human_KEGG$gene_symbol, human_KEGG$gs_name)
四、总结
| 解决方案 | 说明 |
|----------|------|
| 增加超时时间 | 通过 options(timeout = 600) 提高请求等待时间 |
| 使用本地缓存 | 避免重复下载,提高效率 |
| 分页加载 | 适用于大数据量,防止请求失败 |
| 检查网络环境 | 确保能正常访问 MSigDB 服务器 |
| 手动下载基因集 | 完全离线操作,适合复杂网络环境 |
如有其他相关问题(如基因集格式转换、富集分析等),欢迎继续提问!