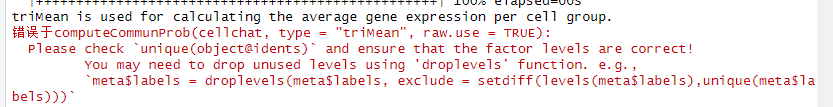
运行细胞通讯分析报错
for(i in names(scRNA.list)){
data.input <- GetAssayData(scRNA.list[[i]], layer = 'data')
meta <- scRNA.list[[i]]@meta.data
# 建议提前对celltype进行排序~
identical(rownames(meta),colnames(data.input))
celltype_order <- c("Unknown",
"Migrating interneuron",
"Satellite glial cell",
"Cortical neuron",
"Endothelial cell",
"Basket cell",
"Ventral otocyst",
"Bergmann glial cell",
"Migrating neuron",
"Mitotic tumor cell",
"Type II spiral ganglion neuron",
"Type I spiral ganglion neuron",
"Cortex layer 6 pyramidal cell",
"Astrocyte",
"Chandelier cell",
"Oligodendrocyte",
"Neuronal progenitor cell",
"Homeostatic microglial cell",
"Type IB spiral ganglion neuron",
"Erythroid terminal differentiation cell",
"Cajal retzius cell")
meta$celltype <- factor(meta$celltype ,levels = celltype_order)
# 根据 meta$celltypes 的顺序进行排序
ordered_indices <- order(meta$celltype)
# 对 meta 和 data.input 进行排序
meta <- meta[ordered_indices, ]
data.input <- data.input[, ordered_indices]
identical(rownames(meta),colnames(data.input))
# 构建cellchat
cellchat <- createCellChat(object = data.input,
meta = meta,
group.by = "celltype")
levels(cellchat@idents)
#设置配体-受体相互作用数据库
CellChatDB <- CellChatDB.mouse # use CellChatDB.mouse if running on mouse data
showDatabaseCategory(CellChatDB)
dplyr::glimpse(CellChatDB$interaction)
# 使用CellChatDB的中特定的数据库进行细胞-细胞通信分析
# 示例中使用了Secreted Signaling
# CellChatDB.use <- subsetDB(CellChatDB, search = "Secreted Signaling", key = "annotation")
# Only uses the Secreted Signaling from CellChatDB v1
# CellChatDB.use <- subsetDB(CellChatDB, search = list(c("Secreted Signaling"), c("CellChatDB v1")), key = c("annotation", "version"))
# 使用所有CellChatDB数据进行细胞-细胞通信分析。
CellChatDB.use <- CellChatDB
# 在构建的cellchat中设定需要使用的数据库
cellchat@DB <- CellChatDB.use
######预处理细胞-细胞通讯分析的表达数据
cellchat <- subsetData(cellchat)
# future::plan("multisession", workers = 1) # do parallel
cellchat <- identifyOverExpressedGenes(cellchat)
cellchat <- identifyOverExpressedInteractions(cellchat)
# 默认情况下,cellchat使用object@data.signaling进行网络推断
# 此外提供了projectData函数,将基因投射到PPI,开发者说PPI并不会或导致很少的伪通讯
cellchat <- projectData(cellchat, PPI.mouse)
#####预测细胞-细胞通信网络
# 该分析的关键参数是类型,即计算每个细胞组的平均基因表达的方法。
# 默认情况下,type = “triMean”,产生较少但更强的交互。
# 当设置 type = “truncatedMean” 时,应对trim设置一个值,进而产生更多交互。
cellchat <- computeCommunProb(cellchat, type = "triMean",raw.use = TRUE)
# 如下
# computeAveExpr(cellchat, features = c("CXCL12","CXCR4"),
# type = "truncatedMean",trim = 0.1)
# min.cells是设置阈值,最小是需要10个细胞参与通讯推断(可以自定义)
cellchat <- filterCommunication(cellchat, min.cells = 10)
#####在信号通路水平上推断细胞间通讯
# CellChat通过汇总与每个信号通路相关的所有配体-受体相互作用的通信概率来计算信号通路水平上的通信概率。
# NB:推断的每个配体-受体对的细胞间通信网络和每个信号通路分别存储在槽'net'和'netP'中。
cellchat <- computeCommunProbPathway(cellchat)
# 计算聚合细胞-细胞通信网络
# 互作网络整合,可以设置soure和target,不设置就是默认全部
cellchat <- aggregateNet(cellchat)
#数据提取,subsetCommunication函数
df.net <- subsetCommunication(cellchat)
saveRDS(cellchat,paste0(i,"_Cellchat.rds"))
}