让【宝码香车】来帮你解答,本回答参考DeepSeek编写,并整理提供。
如果还有疑问可以“评论”。如果答案让您满意,请“采纳”,非常感谢!
这个问题是典型的MCMCglmm系统发育矩阵匹配问题。虽然表面上物种名完全匹配,但实际上可能存在一些隐藏的问题。
问题分析
错误提示表明有些物种在随机效应中出现了,但在ginverse矩阵中没有对应的行。即使你检查了物种名完全匹配,仍可能存在以下问题:
排查步骤
1. 检查数据中的NA值处理
# 更全面的检查
species_in_data <- unique(dat$species)
species_in_data <- species_in_data[!is.na(species_in_data)]
species_in_Ainv <- rownames(Ainv)
# 检查是否有空白字符
cat("数据中物种名示例:\n")
print(head(species_in_data))
cat("Ainv中物种名示例:\n")
print(head(species_in_Ainv))
# 检查字符编码和空白
cat("是否有空白字符:\n")
print(any(grepl("^\\s+|\\s+$", species_in_data)))
print(any(grepl("^\\s+|\\s+$", species_in_Ainv)))
2. 检查随机效应中的实际使用物种
# 查看模型实际使用的物种
library(MCMCglmm)
# 先运行一个简化版检查
test_formula <- update(forms_medium, . ~ . -1) # 移除截距以便诊断
test_model <- try(MCMCglmm(test_formula,
random = ~ species,
data = dat,
verbose = FALSE,
nitt = 100, # 减少迭代次数用于测试
burnin = 10,
thin = 1))
# 如果还是报错,检查随机效应矩阵
species_used <- unique(model.frame(test_formula, data = dat)$species)
species_used <- species_used[!is.na(species_used)]
cat("模型中实际使用的物种数:", length(species_used), "\n")
cat("与Ainv的差异:\n")
print(setdiff(species_used, species_in_Ainv))
3. 系统发育矩阵完整性检查
# 检查Ainv矩阵的完整性
cat("Ainv矩阵维度:", dim(Ainv), "\n")
cat("Ainv是否为方阵:", nrow(Ainv) == ncol(Ainv), "\n")
cat("Ainv对角线是否有零值:", any(diag(Ainv) == 0), "\n")
cat("Ainv是否有NA值:", any(is.na(Ainv)), "\n")
# 检查矩阵是否对称(系统发育矩阵通常对称)
is_symmetric <- all.equal(Ainv, t(Ainv), tolerance = 1e-10)
cat("Ainv是否对称:", is_symmetric, "\n")
4. 数据预处理建议
# 数据清理步骤
dat_clean <- dat
# 1. 去除物种名首尾空格
dat_clean$species <- trimws(as.character(dat_clean$species))
rownames(Ainv) <- trimws(rownames(Ainv))
colnames(Ainv) <- trimws(colnames(Ainv))
# 2. 确保物种名编码一致
dat_clean$species <- iconv(dat_clean$species, to = "UTF-8")
rownames(Ainv) <- iconv(rownames(Ainv), to = "UTF-8")
colnames(Ainv) <- iconv(colnames(Ainv), to = "UTF-8")
# 3. 移除数据中在Ainv中不存在的物种(即使你认为没有)
valid_species <- dat_clean$species %in% rownames(Ainv)
dat_final <- dat_clean[valid_species & !is.na(valid_species), ]
cat("清理后数据行数:", nrow(dat_final), "\n")
cat("清理后物种数:", length(unique(dat_final$species)), "\n")
问题排查流程
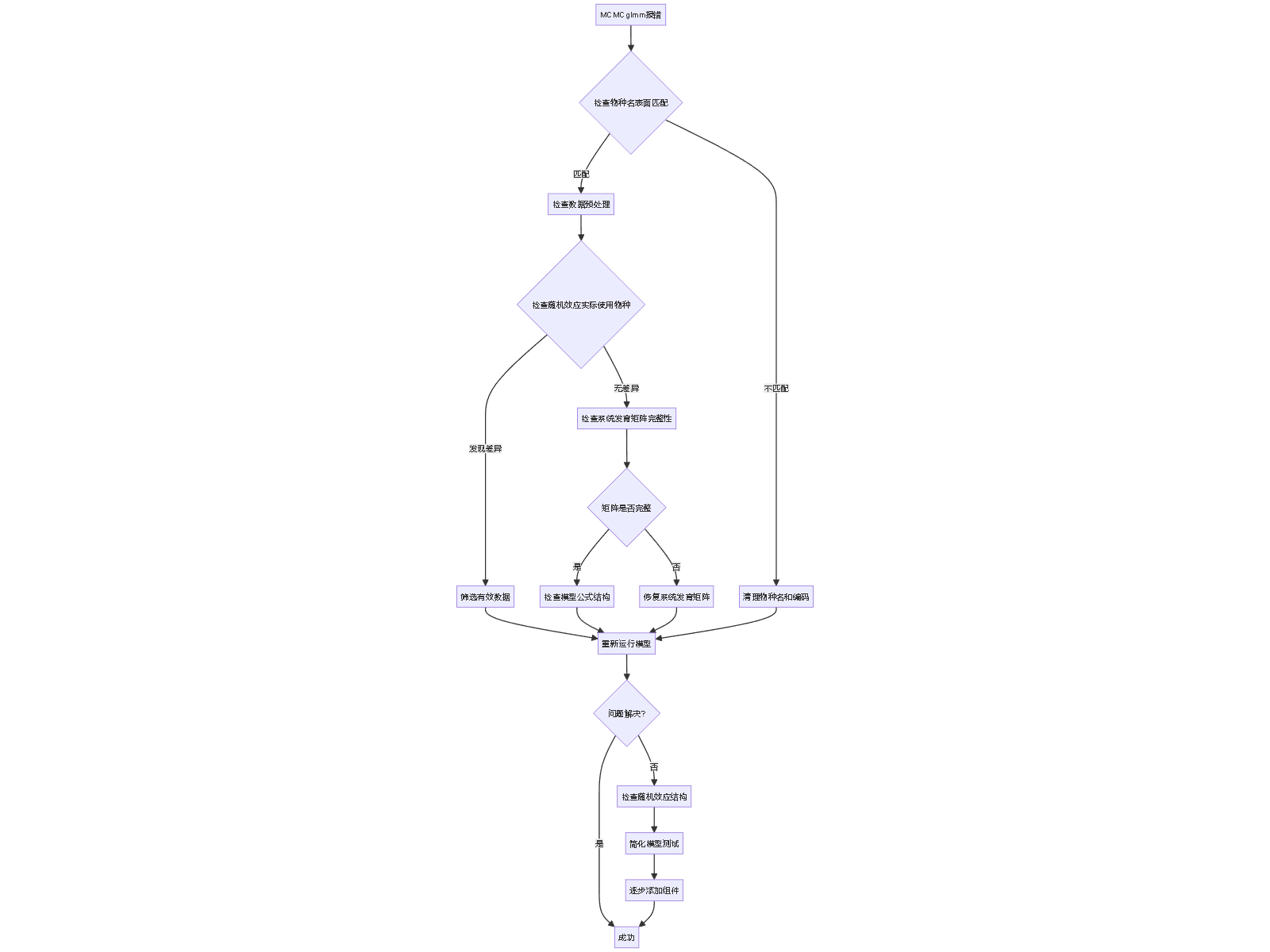
最终解决方案
如果上述步骤仍不能解决问题,尝试以下完整方案:
# 完整的数据准备流程
prepare_phylogenetic_data <- function(data, Ainv_matrix) {
# 清理物种名
data$species <- trimws(as.character(data$species))
rownames(Ainv_matrix) <- trimws(rownames(Ainv_matrix))
colnames(Ainv_matrix) <- trimws(colnames(Ainv_matrix))
# 统一编码
data$species <- iconv(data$species, to = "UTF-8")
rownames(Ainv_matrix) <- iconv(rownames(Ainv_matrix), to = "UTF-8")
colnames(Ainv_matrix) <- iconv(colnames(Ainv_matrix), to = "UTF-8")
# 筛选有效数据
valid_rows <- data$species %in% rownames(Ainv_matrix) & !is.na(data$species)
data_clean <- data[valid_rows, ]
# 确保物种为因子且水平与Ainv一致
data_clean$species <- factor(data_clean$species,
levels = rownames(Ainv_matrix))
return(list(data = data_clean, Ainv = Ainv_matrix))
}
# 使用准备函数
prepared_data <- prepare_phylogenetic_data(dat, Ainv)
# 最终模型运行
final_model <- MCMCglmm(forms_medium,
random = ~ species,
ginverse = list(species = prepared_data$Ainv),
data = prepared_data$data,
family = "gaussian", # 根据你的数据调整
nitt = 13000,
burnin = 3000,
thin = 10)
主要问题通常出现在:
- 隐藏的空白字符
- 字符编码不一致
- 数据中实际使用的物种与检查时的不一致
- 系统发育矩阵不完整或不规范
按照上述流程逐步排查,应该能解决你的问题。








