# install.packages("colorspace")
# options(repos = c(CRAN = "https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
# install.packages("stringi")
# install.packages("ggplot2")
#
# if (!requireNamespace("BiocManager", quietly = TRUE))
# install.packages("BiocManager")
# BiocManager::install("org.Hs.eg.db")
# BiocManager::install("DOSE")
# BiocManager::install("clusterProfiler")
# BiocManager::install("enrichplot")
#引用包
library("clusterProfiler")
library("org.Hs.eg.db")
library("enrichplot")
library("ggplot2")
pvalueFilter = 0.05 #p值过滤条件
qvalueFilter = 0.05 #矫正后的p值过滤条件
#定义颜色
colorSel = "qvalue"
if(qvalueFilter>0.05){
colorSel = "pvalue"
}
setwd("F:\\A基于神经网络的胃癌基因筛选\\3.GO富集分析") #设置工作目录
rt = read.table("diff.txt", header = T, sep = "\t", check.names = F) #读取输入文件
#基因名字转换为基因id
genes = as.vector(rt[,1])
entrezIDs = mget(genes, org.Hs.egSYMBOL2EG, ifnotfound = NA)
entrezIDs = as.character(entrezIDs)
gene = entrezIDs[entrezIDs != "NA"] #去除基因id为NA的基因
#GO富集分析
kk = enrichGO(gene = gene, OrgDb = org.Hs.eg.db, pvalueCutoff = 1, qvalueCutoff = 1, ont = "all", readable = T)
GO = as.data.frame(kk)
GO = GO[(GO$pvalue<pvalueFilter & GO$qvalue<qvalueFilter),]
#保存富集结果
write.table(GO, file = "GO.txt", sep = "\t", quote = F, row.names = F)
#定义显示GO的数目
showNum = 10
if(nrow(GO)<30){
showNum = nrow(GO)
}
#柱状图
pdf(file = "barplot.pdf", width = 7, height = 7)
bar = barplot(kk, drop = TRUE, showCategory = showNum, split = "ONTOLOGY", color = colorSel) + facet_grid(ONTOLOGY~., scale = 'free')
print(bar)
dev.off()
#气泡图
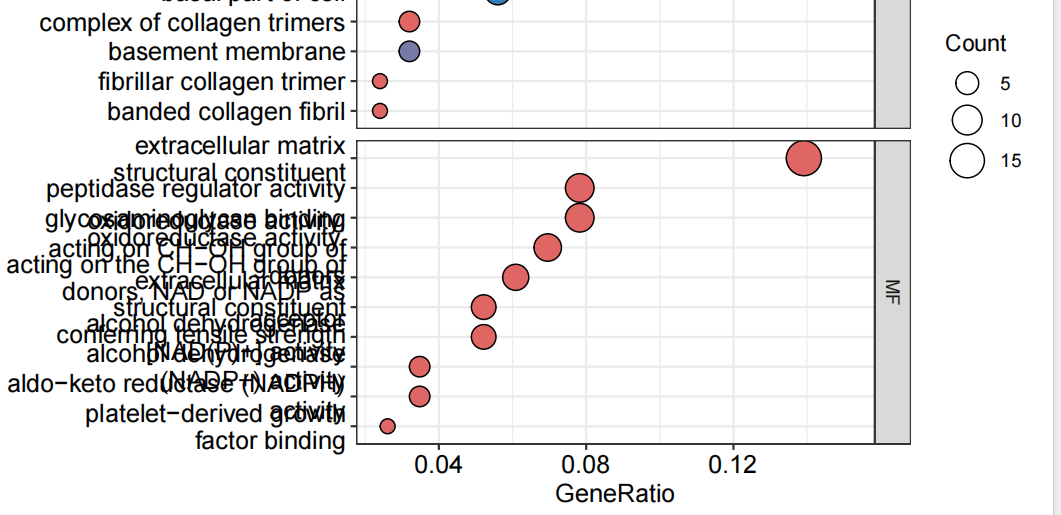
pdf(file = "bubble.pdf", width = 7, height = 7)
bub = dotplot(kk, showCategory = showNum, orderBy = "GeneRatio", split = "ONTOLOGY", color = colorSel) + facet_grid(ONTOLOGY~., scale = 'free')
print(bub)
dev.off()
#获取GO信息
go=data.frame(Category=GO$ONTOLOGY, ID=GO$ID, Term=GO$Description, Genes = gsub("/", ", ", GO$geneID), adj_pval = GO$p.adjust)
#读取基因的logFC
genelist <- data.frame(ID=rt$id, logFC=rt$logFC)
row.names(genelist)=genelist[,1]
#设置圈图参数
circ <- circle_dat(go, genelist)
termNum =8 #显示GO数目
termNum=ifelse(nrow(go)<termNum,nrow(go),termNum)
geneNum=200 #限定基因数目
geneNum=ifelse(nrow(genelist)<geneNum, nrow(genelist), geneNum)
#绘制圈图
chord <- chord_dat(circ, genelist[1:geneNum,], go$Term[1:termNum])
pdf(file="GOcircos.pdf", width=10, height=10)
GOChord(chord,
space = 0.001, #基因之间的间距
gene.order = 'logFC', #按照logFC值对基因排序
gene.space = 0.25, #基因名跟圆圈的相对距离
gene.size = 5, #基因名字体大小
border.size = 0.1, #线条粗细
process.label = 6) #GO字体大小
dev.off()
#聚类图
pdf(file="GOcluster.pdf",width=12, height=10)
GOCluster(circ,
go$Term[1:termNum],
lfc.space = 0.2, #logFC与树之间的空隙大小
lfc.width = 1, #logFC的圆圈宽度
term.space = 0.2, #logFC与GO之间空隙的大小
term.width = 1) #GO圆圈的宽度
dev.off()








